权利要求
1.一种PI纤维基原位
复合材料的制备方法,其特征在于,包括如下步骤:
S1、将对苯二胺粉末溶解于溶剂中,搅拌10-60min,得到ODA溶液;
S2、在步骤S1得到的ODA溶液中加入均苯四甲酸酐,持续搅拌,得到PAA溶液;
S3、在步骤S2得到的PAA溶液中加入压电
陶瓷粉体,通过静电纺丝将加入压电陶瓷粉体的PAA混合溶液制成PAA复合纳米纤维膜,经梯度加热处理得到PI纳米纤维膜;
S4、将
锂盐和助剂按质量比(0.2-1.5):(0.5-4)混合均匀,得到原位聚合前驱体溶液;
S5、将步骤S4中得到的原位聚合前驱体溶液滴加到步骤S3中得到PI纳米纤维膜上,静置1h,得到PI纤维基原位复合材料。
2.根据权利要求1所述的制备方法,其特征在于,步骤S1中所述溶剂为N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、N-甲基吡咯烷酮中的一种。
3.根据权利要求1所述的制备方法,其特征在于,步骤S3中所述压电陶瓷粉体粒径为50nm-500nm的BaTiO3,Nb2O5,CeO2,Bi4Ti3O12,KNN,PZT中的一种。
4.根据权利要求1所述的制备方法,其特征在于,步骤S3中所述静电纺丝条件为在15-100 kV的高压静电场中纺丝,注射器推进速度为10-1000 mL/h,纺丝间距10-80 cm,滚筒转速20-100 rpm。
5.根据权利要求1所述的制备方法,其特征在于,步骤S3中所述梯度加热处理条件包括,将PAA复合纳米纤维膜在100℃下干燥1-2h,升温,在5-10℃/min的加热速率下,分别在160℃、250℃和300℃加热0.5-2h。
6.根据权利要求1所述的制备方法,其特征在于,步骤S4中所述锂盐为四氟硼酸锂、高氯酸锂、三氟甲磺酸锂、双三氟甲磺酰亚胺锂、二氟草酸硼酸锂和
六氟磷酸锂中一种或多种。
7.根据权利要求1所述的制备方法,其特征在于,步骤S4中助剂包括助溶剂和/或引发剂,助溶剂和引发剂的质量比为(1.8-2.2):(0.01-0.02)。
8.根据权利要求7所述的制备方法,其特征在于,所述助溶剂为碳酸乙烯酯、碳酸二甲酯、碳酸二乙酯、碳酸甲乙酯、碳酸二丙酯、碳酸丙烯酯和乙二醇二甲醚中一种或多种;引发剂为偶氮二异丁腈、偶氮二异庚腈或偶氮二异丁酸二甲酯一种或多种。
9.如权利要求1-8任一项所述制备方法制备所得的PI纤维基原位复合材料。
10.如权利要求1-8任一项所述制备方法制备所得的PI纤维基原位复合材料或权利要求9所述的PI纤维基原位复合材料在固态电解质中的应用。
说明书
技术领域
[0001]本发明属于复合材料制备技术领域,具体涉及一种PI纤维基原位复合材料及其制备方法和应用。
背景技术
[0002]随着现代电子设备向小型化、轻量化和高性能化方向发展,对能源存储系统的要求也越来越高。特别是在移动设备、电动汽车和智能穿戴等领域,对电池性能的需求不断增长。传统的液态电解质锂离子电池虽然已广泛应用于各个领域,但存在泄漏、易燃和热稳定性差等问题,限制了其在更广泛高温或极端环境下的应用。
[0003]为了解决这些问题,固态电解质应运而生。固态电解质由于其不可燃性和高热稳定性,被认为是提高电池安全性和能量密度的有效途径。然而,现有的固态电解质材料仍存在一些技术瓶颈,如离子电导率较低、与电极材料的界面兼容性差、制备工艺复杂等。例如,专利CN202010249181公开了一种复合凝胶固态电解质及其制备方法和全固态锂离子电池,但制备流程较为苛刻,需要使用真空反应釜和超临界成型装置。专利CN202110960082公开了一种通过冷压复合多层材料制备的聚合物固态电解质,但缺少无机填料的添加,导致锂离子迁移率较低,热稳定性不足。专利CN116344928A公开了一种以玻璃纤维作为载体的锂离子电池凝胶电解质的原位构筑方法,但玻璃纤维的拉伸力学性能较差,限制了其在柔性电子设备中的应用。专利CN202410124905.3涉及一种高性能复合聚合物电解质膜的制备及其应用,但采用的非原位聚合方法导致凝胶电解质和基体的结合能力较差,影响了电解质的整体性能。
[0004]综上所述,现有技术在固态电解质的离子电导率、热机械稳定性、制备工艺简便性以及与电极材料的兼容性等方面仍有待进一步提高。因此,开发具有高离子电导率、优异热机械性能以及简便制备工艺的新型固态电解质材料,对于推动全
固态电池技术的发展具有重要意义。
发明内容
[0005]本发明的目的是提供一种PI纤维基原位复合材料及其制备方法和应用,该PI纤维基原位复合材料具有高离子电导率和优异热机械性能。
[0006]为解决上述技术问题,本发明采用的技术方案如下:
一种PI纤维基原位复合材料的制备方法,包括如下步骤:
S1、将对苯二胺粉末溶解于溶剂中,搅拌10-60min,得到ODA溶液;
S2、在步骤S1得到的ODA溶液中加入均苯四甲酸酐,持续搅拌,得到PAA溶液;
S3、在步骤S2得到的PAA溶液中加入压电陶瓷粉体,通过静电纺丝将上述混合溶液制成PAA复合纳米纤维膜,经梯度加热处理得到PI纳米纤维膜;
S4、将锂盐和助剂按质量比(0.2-1.5):(0.5-4)混合均匀,得到原位聚合前驱体溶液;
S5、将步骤S4中得到的原位聚合前驱体溶液滴加到步骤S3中得到PI纳米纤维膜上,静置1h,得到PI纤维基原位复合材料。
[0007]优选的,步骤S1中将对苯二胺粉末以10-20wt%的重量百分比溶解于溶剂中,搅拌10-60min,得到ODA溶液。
[0008]优选的,步骤S1中所述溶解苯二胺粉末的溶剂为N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、N-甲基吡咯烷酮中的一种。
[0009]优选的,步骤S3中所述压电陶瓷粉体粒径为50nm-500nm的BaTiO3,Nb2O5,CeO2,Bi4Ti3O12,KNN,PZT中的一种。
[0010]优选的,步骤S3中所述静电纺丝条件为在15-100 kV的高压静电场中纺丝,注射器推进速度为10-1000 mL/h,纺丝间距10-80 cm,滚筒转速20-100 rpm。
[0011]优选的,步骤S3中所述梯度加热处理条件包括,将PAA复合纳米纤维膜在100℃下干燥1-2h,升温,在5-10℃/min的加热速率下,分别在160℃、250℃和300℃加热0.5-2h。
[0012]优选的,步骤S4中所述锂盐为四氟硼酸锂、高氯酸锂、三氟甲磺酸锂、双三氟甲磺酰亚胺锂、二氟草酸硼酸锂和六氟磷酸锂中一种或多种;
优选的,步骤S4中助剂包括助溶剂和/或引发剂,助溶剂和引发剂的质量比为(1.8-2.2):(0.01-0.02)。
[0013]优选的,所述助溶剂为碳酸乙烯酯、碳酸二甲酯、碳酸二乙酯、碳酸甲乙酯、碳酸二丙酯、碳酸丙烯酯和乙二醇二甲醚中一种或多种;引发剂为偶氮二异丁腈、偶氮二异庚腈或偶氮二异丁酸二甲酯一种或多种。
[0014]本发明还提供了一种如所述制备方法制备所得的PI纤维基原位复合材料。
[0015]本发明还提供了一种如所述制备方法制备所得的PI纤维基原位复合材料或所述的PI纤维基原位复合材料在固态电解质中的应用。
[0016]本发明采用静电纺丝技术,结合ODA和PMDA的化学反应制备PAA溶液,在PAA溶液中,引入了粒径为50nm-500nm的压电陶瓷粉体,以增强材料的介电性能。通过梯度加热,将PAA复合纳米纤维膜转化为黄色的PI纳米纤维膜,确保纳米纤维膜的高强度和柔性。本发明通过精确配比锂盐、助剂(助溶剂和引发剂),形成化学性质稳定的前驱体溶液,通过严格的比例控制实现PI纤维基原位复合材料的高离子电导率和化学稳定性。通过将前驱体溶液滴加到PI纳米纤维膜上并静置,利用原位聚合反应在膜内形成均匀的PI纤维基原位复合材料,这种原位聚合技术不仅确保了复合材料的结构均匀性,而且赋予了材料优异的综合性能,包括高电导率、高机械强度和良好的热稳定性,满足了高性能固态电解质的需求。
[0017]与现有技术相比,本发明具有如下优点和技术效果:
1.本发明的PI纤维基原位复合材料,在室温条件下展现出1-5×10-4S/cm的高离子电导率,显著增强电池的导电性能,更高效地进行离子传输,在充放电过程中实现更快的响应速度和更高的能量转换效率,有助于提高电池的整体性能。
[0018]2.本发明的PI纤维基原位复合材料机械强度高达31 MPa,高机械强度使复合材料能够承受日常使用中的物理冲击和压力,延长电池的使用寿命,确保其在各种应用场景下的结构稳定性。
[0019]3.本发明的PI纤维基原位复合材料在100-160℃温度内,展现出卓越的耐热性,使得电池即使在高温或极端环境下也能保持稳定工作,拓宽其应用范围,特别是在高温工业应用或户外设备中,确保了电池的安全性和稳定性。
[0020]4.本发明通过引入PI纳米纤维膜,使复合材料具备良好的柔性,能够适应可穿戴设备和柔性电子设备的需求,柔性特性为电池设计提供了更大的灵活性,电池能够与复杂形状的设备表面贴合,同时保持其
电化学性能。
[0021]下面通过附图和实施例,对本发明的技术方案做进一步的详细描述。
附图说明
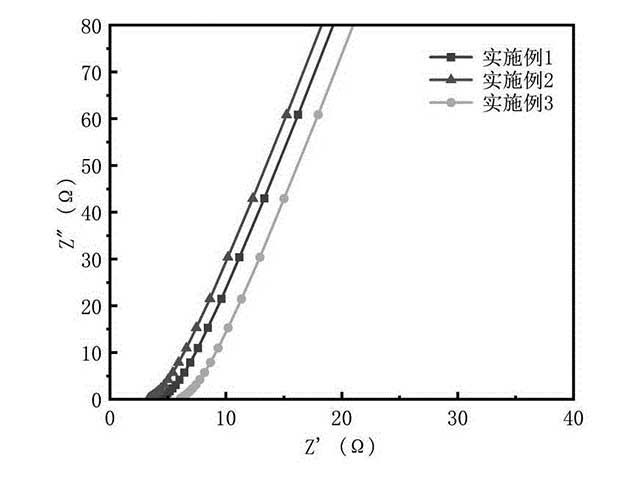
[0022]图1为本发明实施例1-3提供的PI纤维基原位复合材料电导率。
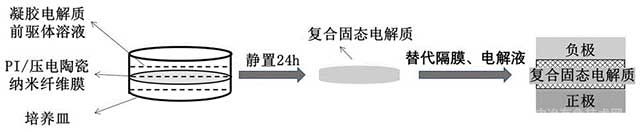
[0023]图2为本发明PI纤维基原位复合固态电解质的制备流程示意图。
[0024]图3为本发明实施例1提供的PI纤维基原位复合材料的扫描电镜图。
具体实施方式
[0025]以下通过附图和实施例对本发明的技术方案做进一步说明。
[0026]除非另外定义,本发明使用的技术术语或者科学术语应当为本发明所属领域内具有一般技能的人士所理解的通常意义。
[0027]在本发明中,若无特殊说明,其他试验材料及仪器设备均为本领域常规的试验材料,均可通过商业渠道购买得到。
[0028]实施例1 本实施提供了一种PI纤维基原位复合材料,制备方法包括如下步骤:
S1、将1.63 mol对苯二胺粉末溶解于1000 mL N,N-二甲基甲酰胺中,搅拌10min,得到ODA溶液;
S2、在步骤S1得到的ODA溶液中加入0.88 mol的均苯四甲酸酐,持续搅拌,得到PAA溶液;
S3、在步骤S2得到的PAA溶液中加入0.43 mol粒径为50nm BaTiO3,通过在15 kV的高压静电场中纺丝,注射器推进速度为10 mL/h,纺丝间距15 cm,滚筒转速50 rpm将上述混合溶液制成PAA复合纳米纤维膜,将PAA复合纳米纤维膜在100℃下干燥1h,升温,在5℃/min的加热速率下,分别在160℃、250℃和300℃加热0.5h得到PI纳米纤维膜;
S4、将100 g四氟硼酸锂、100 g碳酸乙烯酯和1 g偶氮二异丁腈混合均匀,得到原位聚合前驱体溶液;
S5、将步骤S4中得到的原位聚合前驱体溶液滴加到步骤S3中得到PI纳米纤维膜上,静置1h,得到PI纤维基原位复合材料。
[0029]实施例2 本实施提供了一种PI纤维基原位复合材料,制备方法包括如下步骤:
S1、将1.63 mol对苯二胺粉末溶解于1000 mL N,N-二甲基乙酰胺中,搅拌30min,得到ODA溶液;
S2、在步骤S1得到的ODA溶液中加入0.88 mol的均苯四甲酸酐,持续搅拌,得到PAA溶液;
S3、在步骤S2得到的PAA溶液中加入0.38 mol粒径为250nm Nb2O5,通过在15 kV的高压静电场中纺丝,注射器推进速度为10 mL/h,纺丝间距15 cm,滚筒转速50 rpm,将上述混合溶液制成PAA复合纳米纤维膜,将PAA复合纳米纤维膜在100℃下干燥1h,升温,在7.5℃/min的加热速率下,分别在160℃、250℃和300℃加热1h得到PI纳米纤维膜;
S4、将100 g高氯酸锂、100 g碳酸二甲酯和1 g偶氮二异庚腈混合均匀,得到原位聚合前驱体溶液;
S5、将步骤S4中得到的原位聚合前驱体溶液滴加到步骤S3中得到PI纳米纤维膜上,静置1h,得到PI纤维基原位复合材料。
[0030]实施例3 本实施提供了一种PI纤维基原位复合材料,制备方法包括如下步骤:
S1、将1.63 mol对苯二胺粉末溶解于1000 mL N,N-二甲基乙酰胺中,搅拌60min,得到ODA溶液;
S2、在步骤S1得到的ODA溶液中加入0.88 mol的均苯四甲酸酐,持续搅拌,得到PAA溶液;
S3、在步骤S2得到的PAA溶液中加入0.58 mol粒径为500nm CeO2,通过在15 kV的高压静电场中纺丝,注射器推进速度为10 mL/h,纺丝间距15 cm,滚筒转速50 rpm,将上述混合溶液制成PAA复合纳米纤维膜,将PAA复合纳米纤维膜在100℃下干燥1h,升温,在10℃/min的加热速率下,分别在160℃、250℃和300℃加热2h得到PI纳米纤维膜;
S4、将100 g三氟甲磺酸锂、100 g碳酸二乙酯和1 g偶氮二异丁酸二甲酯混合均匀,得到原位聚合前驱体溶液;
S5、将步骤S4中得到的原位聚合前驱体溶液滴加到步骤S3中得到PI纳米纤维膜上,静置1h,得到PI纤维基原位复合材料。
[0031]通过以下试验对上述实施例1-3提供的PI纤维基原位复合材料进行效果验证,试验方案如下:
将实施例1-3制备得到的PI纤维基原位复合材料用冲击孔冲击得到电解质膜圆片(膜直径为16mm),进行电导率测试,测试方法如下:将电解质膜圆片组装成CR2032纽扣电池,按照正极、不锈钢片、电解质膜、不锈钢片、负极的顺序进行组装,设置交流阻抗振幅电压为5mV,频率范围10mHz-100KHz(电化学工作站),再进行EIS的测试,得到电化学阻抗数据。
[0032]对于得到的电化学阻抗数据,通过Zview软件进行等效电路拟合,得到实施例1-3制备得到的PI纤维基原位复合材料的总电阻。根据上述样品的厚度和电极的面积等参数,计算得到样品的总电导率。结果如图1。
[0033]由图1可知,实施例1-3制备得到的PI纤维基原位复合材料的电导率均高于1mS/cm,明显优于目前
锂电池常用的
隔膜加液态电解质的电导率(0.6mS/cm)。
[0034]对实施例1提供的PI纤维基原位复合材料进行电镜扫描,结果如图3。
[0035]由图3可知,通过在自支撑、柔性PI纤维素骨架表面原位聚合形成的PI纤维基复合材料具有完整的表面,平坦的界面。在用作固态电解质时,这种原位聚合的平整界面很大程度上有助于降低固态电解质层的体积电阻。
[0036]最后应说明的是:以上实施例仅用以说明本发明的技术方案而非对其进行限制,尽管参照较佳实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对本发明的技术方案进行修改或者等同替换,而这些修改或者等同替换亦不能使修改后的技术方案脱离本发明技术方案的精神和范围。
说明书附图(3)

声明:
“PI纤维基原位复合材料及其制备方法和应用” 该技术专利(论文)所有权利归属于技术(论文)所有人。仅供学习研究,如用于商业用途,请联系该技术所有人。
我是此专利(论文)的发明人(作者)
 1116
编辑:北方有色网
来源:浙江柔荷新能源材料有限公司
1116
编辑:北方有色网
来源:浙江柔荷新能源材料有限公司



 咨询细节
咨询细节









































































 2026年08月06日 ~ 08日
2026年08月06日 ~ 08日 















