近年来,各类金属纳米催化剂及其催化研究已取得了较大进展,但是单金属催化剂仍然存在一些问题,譬如单金属催化剂活性位单一、在高温下易烧结、易被毒物竞争吸附以及对抗CO2和耐SO2性能较差(即在CO2或SO2存在条件下催化活性显著下降)等
因此,很有必要建立一种全新方法,合成具有特定结构的催化剂,既能稳定催化活性中心,又能构筑新活性位且能改善抗CO2和耐SO2性能
目前,人们已进行广泛深入的相关研究
其中制备双金属催化剂是一种较为有效的策略
双金属催化剂中由于第二种金属的引入,除金属与载体之间存在相互作用外,两种金属之间也会产生相互作用,因而可改变催化剂活性组分的结构,稳定活性中心
由于某些双金属催化剂中的双金属纳米粒子具有特殊的电子效应和表面结构,因此双金属催化剂对加氢、裂解等反应表现出优良的活性和选择性,并应用于多种化工生产过程
在双金属系统中,金属存在状态主要有两种:合金[1]和异质结[2,3](核壳、异质等结构)
根据合金的结构,可将之分为两类,即固溶合金和金属间化合物
前者通常是由具有相似原子尺寸和电子特性的金属替代另一种金属而形成的合金
这种随机原子排列的结构与原来的金属结构相同(图1a)
若一种元素的原子小到可进入母金属的晶格空隙,则可形成间隙固溶合金(图1b)
与固溶合金相对的则为金属间化合物
金属间化合物为两种金属元素呈规律性排布,形成高度有序原子排列的晶体结构(图1c)
通常,两种金属的相似性可通过其在元素周期表中相对位置进行粗略估计[4]
目前,以固溶合金为催化剂的研究报道较多,由于固溶合金的原子排列结构较为随机,因此本文综述了近年来国内外学者对原子排列结构更为有序的金属间化合物的合成及其催化性能
1 金属间化合物的合成方法
从理论上讲,金属间化合物可由元素周期表中的各个金属元素(甚至是一些非金属元素)自由组合而成
但若要将金属间化合物用于催化领域,则其必须含有一个作为活性中心的元素(如Pd、Pt、Au、Ru、Rh、Ir等
贵金属以及Mn、Fe、Co、Ni、Cu等过渡金属元素)
另外,从制备的难易程度来看,目前金属间化合物较为常用的第二种元素主要是半导体及其相近元素(如Ga、Ge、Sb、Sn、Bi、Pb、In、Al等)、前过渡金属元素(如Ti、V、Zr等)以及一些主族元素(如Na、K、Mg等碱金属或碱土金属等)
除此以外,对于某些具有特殊应用的催化剂,为实现双功能的目的,也采用 Zn、Fe、Co、Ni、Cu(甚至Pd、Pt等贵金属)等具有活性的过渡金属元素作为第二种元素来构筑金属间化合物
不同组合的金属间化合物的制备方法大相径庭
例如,采用传统的氢还原法即可容易地制备出排列有序、晶相均一的贵金属与半导体元素组成的金属间化合物纳米粒子[5]
这些制备方法各有优缺点,现总结如下
1.1 化学还原法
化学还原法是指先将金属前驱体溶解在适当的溶剂中,在柠檬酸盐、烷基硫醇或硫代醚等稳定剂存在下,然后利用硼氢化钠、硼烷叔丁胺、氢气等还原剂使金属前驱体还原,从而形成金属间化合物
为控制制备的纳米粒子的直径,反应溶液中也可加入不同聚合物配体(如聚乙烯吡咯烷酮(PVP)、聚乙烯醇(PVA)等)阻止生成的纳米粒子发生团聚,通过调控聚合物配体浓度即可达到粒径可控的目的[6,7,8]
迄今为止,采用化学还原法合成金属间化合物的研究也已有一些报道
例如,Chou和Schaak等[9]以PVP和2-乙基-2-恶唑啉为稳定剂,以NaBH4为还原剂,在四乙二醇溶剂中合成了Sn基双金属间化合物M-Sn(M=Fe,Co,Ni,Pd)
DiSalvo及其合作者采用以NaBH4或萘酸钠为还原剂、以四氢呋喃为溶剂的还原法合成了PtPb和PtBi双金属间化合物纳米晶[10,11]
我们课题组采用以乙酰丙酮钯和氯化镓为前驱体、以硼烷叔丁胺为还原剂的还原法在油胺溶剂中合成了Pd5Ga3金属间化合物纳米晶[12]
虽然从原理和操作条件上看化学还原法很简单,但因为两种金属的化学还原电势差异很大,因而很难控制两种金属同时成核
若使用的还原剂的还原性很强,还原过程非常快,则不易控制两种金属形成金属间化合物
比如,Sra等[13]使用NaBH4为还原剂时发现,金和
铜的还原很快,两者的成核单独发生,没有形成合金
另外,由于金属间化合物中两种金属的原子半径相差较大,晶核难以生长成有序均匀的晶体,故其往往形成合金而非金属间化合物
例如,Li等[14]在以联氨为还原剂合成Ni-Co和Ni-Cu合金时观察到,虽然形成了
镍钴和镍
铜合金,但是还原过程中的晶核生长难以控制,并未形成金属间化合物,且纳米晶的粒径也无法控制
对此,目前主要的解决办法是加入其它稳定剂(如PVP、PVA等)或采用较为温和的还原剂
Liu等[15]以Ni(CH3COO)2·4H2O和Ga(acac)3为前驱体,以甲硼烷-叔丁胺络合物(TBAB)为还原剂,在油胺溶剂中合成了Ni3Ga金属间化合物纳米晶;以NiCl2和SnCl2为前驱体,以正丁基
锂为还原剂,在油胺溶剂中合成了Ni3Sn2金属间化合物
Wu等[16]使用硼烷-叔丁胺络合物(TBAB)为还原剂,以十六烷二醇为稳定剂,获得了Pt3Ni金属间化合物
1.2 沉积沉淀还原法
沉积沉淀法是指先将混合的金属前驱体分解,所形成的复合物沉积(浸渍、沉淀等)到载体上,再将催化剂暴露在氢气等还原性气氛和高温条件下,使催化剂表面上的复合物还原
沉积到载体上的复合物会在氢气作用下实现双金属的重新排列,生成稳定的双金属乃至金属间化合物
例如,Stassi等[17]采用浸渍法将PtFe 或PtSn的氯化物前驱体负载到不同碳载体(CV、CN和CN-P)上,干燥后在氢气气氛中于350oC还原3 h,制得PtFe或PtSn金属间化合物催化剂
Furukawa等[18]采用浸渍法先将Pt和Co前驱体负载到不同载体上,然后在一定温度(如600 oC或800oC,还原温度可视不同载体而定)下于氢气气氛中还原2 h,获得Pt3Co/MOx(M=La,Ca,Mg,Al,Si)的负载型金属间化合物
Komatsu等[19]同样采用等体积浸渍法将金属前驱体负载到Al2O3上,并在800oC和氢气气氛中进行还原,合成了Al2O3负载的PtCu金属间化合物催化剂
值得注意的是,对于贵金属基金属间化合物,当采用沉积沉淀法时,可先浸渍贵金属,再沉积另一种金属,这样仍可合成出金属间化合物
这是由于贵金属对氢气具有活化作用,活化后的氢通过溢流会促使第二种金属化合物的还原,有利于两种金属的充分接触,从而形成金属间化合物
例如,Furukawa等[20]先采用等体积浸渍法制备了Pd/SiO2,然后以Zn(NO3)2(或Ge、In、Sn、Zn的硝酸盐)为前驱体继续浸渍在Pd/SiO2上,最后在800oC还原2 h,制得PdmMn/SiO2 (M=Ge,In,Sn,Zn)金属间化合物催化剂
除上述的浸渍法沉积之外,也有研究者采用沉淀法将前驱体从溶液中沉淀出来,再经过还原和焙烧形成金属间化合物
例如,Ota等[21]通过调控混合前驱体溶液的pH值,使金属产生沉淀,所得沉淀物经焙烧和还原过程,可制得二元或三元金属间化合物
沉积沉淀还原法虽然操作较为简单,但利用氢气在高温条件下还原前驱体的操作具有一定的危险,因而不太适于大规模工业化制备
除此以外,在高温条件下采用氢气还原的过程可能会破坏某些氧化物载体的结构和改变表面元素价态等,从而对催化剂的活性产生不利影响
1.3 电弧熔融法
电弧熔融法是根据电弧放电的原理在电弧熔炉中在很短的距离上施加很高的电压,造成击穿发电,产生的高温使金属达到熔融态
在熔融状态下,各类金属会进行再排列,从而形成金属间化合物
电弧熔融法主要有以下特点:(1)熔融温度可达2000oC左右,可熔化大部分难熔金属;(2)熔融时间为30~60 s,可最大限度地减少元素挥发;(3)猛烈的电弧打火使熔样完全搅拌,匀质性极好;(4)可采用高纯的惰性气体(如氩气等)保护熔样,避免结构和组成变化
到目前为止,已有一些采用电弧熔融法合成金属间化合物的研究报道[22,23,24]
需要注意的是,电弧熔融法为高温熔融状态下金属的再排列,因而所制得的金属间化合物的粒径较大且难以控制,通常需要采用物理破碎的方法才能使其粒径达到一定的尺寸;除此以外,电弧熔融法的能耗较大,难以大规模应用
1.4 化学气相沉积法
采用化学气相沉积法(CVD)合成金属间化合物通常是将第二种金属的前驱体以蒸气形式直接送入母粒金属,随后还原沉积和合金化
由于在适当温度下还原沉积会优先发生在活性金属表面,而不是在惰性载体上,因而可进行选择性沉积,实现高效定向制备金属间化合物
这种方法为了使第二种金属能够以蒸气形式通入反应器中,需要寻找具有足够高的蒸气压的前驱体
较为常用的前驱体包括
硅烷[25]和四烷基化合物[26]等
由此可看出,当采用CVD法制备金属间化合物时,加载的第二种金属的量在很大程度上取决于温度和时间
若要控制第二种金属的加量则需要具备合适的条件,且由于蒸汽化的限制,因而第二种金属的选择受到一定程度的限制
采用CVD法合成金属间化合物的研究已有文献报道
例如,Komatsu等[27]先通过离子交换法将Pt引入MCM-41的骨架中,在200oC还原1 h后得到Pt/MCM-41,随后在150oC下以氢气为载气,将Ge(CH3)4以蒸气形式引入Pt/MCM-41中,最后600oC和氢气存在的条件下还原1 h,即可合成出Pt-Ge金属间化合物
Chen等[25]则以Ni为母金属,在氢气气氛中与蒸气态硅烷反应,合成了不同Ni/Si比例的金属间化合物(Ni2Si、NiSi和NiSi2)
采用化学气相沉积法通常可获得高纯的金属间化合物,且能够控制颗粒的直径和分散性,但其合成条件较为苛刻且产量较低,故难以规模化合成金属间化合物
1.5 多元醇法
多元醇法是一种较为常见的在液相中合成金属间化合物的方法
此种方法的核心在于两种金属在低温下诱导相互扩散和成核,以粉末形式在液相中退火而形成
相对电弧熔融法等无溶剂的方法,多元醇法是在液相中成核与扩散,反应温度较低,且其在反应过程中可在液相中加入聚合物稳定形成的粒子
因此,多元醇法所制得的金属间化合物具有粒径小、粒径可控、不易烧结等优点
较为常用的多元醇有乙二醇、丙二醇和甘油等[28,29]
值得指出的是,在多元醇法中,多元醇的用途不仅是溶剂,也可作为封端剂和还原剂,而且多元醇法不仅可在纳米颗粒上生长粒子,也可在溶胶上进行反应
其反应机理示意如图2所示
图1
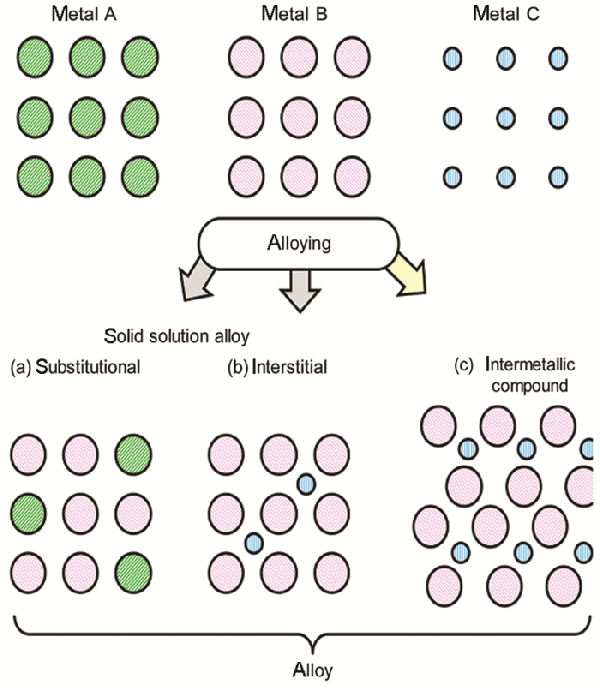
图1(a) 置换固溶合金、(b) 间隙固溶合金和(c) 金属间化合物的结构示意图[4]
Fig.1Structural scheme of (a) substituted solid-solution alloy, (b) interstitial solid-solution alloy, and (c) intermetallic compound[4]
图2
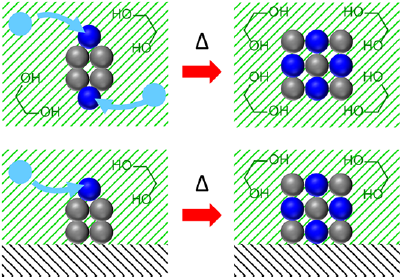
图2采用多元醇法合成金属间化合物的反应机理示意图[4]
Fig.2Schematic illustration of reaction mechanisms for preparation of intermetallic compounds by the polyol method [4]
Schaak及合作者[30,31]以三乙二醇为溶剂,以硼氢化钠为还原剂,以PVP为稳定剂,在不同温度下反应不同时间合成了Ag4Sn、Au5Sn、FeSn2、Ni3Sn4、PtBi、PtPb、PtSb、PtSn等多种金属间化合物
除了上述这些合成方法之外,目前尚有其它方法可用来合成金属间化合物
例如,以金属有机化合物为前驱体经高温热解来合成双金属的热分解法[32,33,34];传统冶金采用的热退火法[35,36];以及采用超声波辅助的超声化学法[37,38]等
由于这些合成方法各有优缺点,因此需要根据实际应用来选用适宜方法合成金属间化合物
2 金属间化合物在催化领域中的应用
在介绍了金属间化合物的各种合成方法后,下面简要总结金属间化合物在催化氧化、催化加氢以及催化重整等反应中的应用
对于这些特定的化学反应,由于催化活性中心不同以及催化机理各异,故要根据不同反应的作用机理来选择、设计不同的金属间化合物
另外,由于催化反应的条件不同,使反应过程中金属间化合物的存在状态及形式也有差异,因此必须考虑催化剂本身的变化来揭示不同反应的催化机理
2.1 氧化反应
正如上述合成方法中所提到的,金属间化合物的合成过程大都需要还原或惰性气氛条件,故而金属间化合物中的金属均为金属态
而对于氧化反应,反应氛围为富氧条件
因而氧化反应中金属间化合物往往会出现相分离,形成两种金属的氧化物混合晶相,这种新形成的混合晶相可能是氧化反应的活性位
除此以外,由于各种金属氧化的难易程度不同,因而金属间化合物在氧化过程中会出现偏析、部分氧化等情况,从而形成具有不同结构(比如核壳、异质结、固溶体等)的金属氧化物,而这些特殊结构有利于提高催化剂的稳定性、抗中毒化性能等
下面以CO氧化为例来说明金属间化合物在氧化反应中的催化应用
CO氧化因其氧化过程简单而常用于探针反应
CO氧化对基础化学研究、汽车尾气净化以及燃料电池等研究均具有重要的意义
在这些研究领域中,Au、Pt、Pd或Rh基催化剂为重点研究的催化材料[39,40,41,42],尤其是在这些催化材料的(110)晶面上的研究较多
基于此,众多研究者从Au、Pt、Pd或Rh基的金属间化合物催化剂入手,探讨金属间化合物对CO氧化的催化作用
例如,Saravanan等[43]制备了用于CO氧化的Pt3Ti/SiO2催化剂,发现与Pt/SiO2相比,前者表现出优异的催化性能:与负载单Pt催化剂对比,Pt3Ti/SiO2催化剂上的CO转化率提高了三倍,且起燃温度下降了大约80oC
作者认为,这是由于CO在Pt3Ti上的吸附弱于在Pt上的(TPD结果显示),从而减小了CO对催化剂的毒化作用
Komatsu等[26]比较了各种Pt基金属间化合物催化剂(PtmMn/SiO2;M=Co,Cu,Fe,Ge,Sn,Tl)对CO氧化的催化活性后发现,Pt3Co和PtCu显示最优的催化活性
利用IR-TPD联用技术发现,在氧化过程中Cu富集到PtCu表面;CO表面在铜上的吸附作用明显弱于在铂上的[19]
此外,对CO氧化的动力学研究结果显示,O2与CO存在竞争吸附,铜的引入促进了O2的吸附
这些结果表明,铜的引入促进了O2的吸附,抑制了CO的毒化作用,从而提高了催化剂的活性
众所周知,Au对于CO氧化具有最优的催化活性
广大研究者对金基金属间化合物催化剂也开展了广泛而深入的研究
例如,Liu等[44]制备了Au3Cu金属间化合物,并将其负载到SBA-15上,用于CO催化氧化,实现了在室温条件下将CO完全氧化为CO2,而单金属Au和Cu催化剂则显示较低的催化活性
他们利用原位XRD、EPR、XANES以及FTIR等表征技术证实了催化剂的表面为Au和CuOx的混合晶相,Au与Cu之间存在协同作用,CuOx作为一种有效的氧气活化中心和Au作为双活性位促使CO在低温氧化为CO2
除了AuCu金属间化合物,Xiao等[45]制备了碱金属为第二金属的Au2Na金属间化合物,实现了近室温氧化CO(表观活化能为31.6 kJ/mol)
作者通过DFT计算后发现,催化剂暴露到氧气中后,第二层的Na迁移到表面成为氧气的结合位点;随后CO直接与O2反应形成OOCO中间体,其动力学反应途径如图3所示
图3

图3NaAu2(111)和Au(221)晶面上共吸附CO和O2后的反应途径示意图,上、下分别为Au(221)和NaAu2(111)晶面上的反应途径[45]
Fig.3Reaction pathways of CO and O2 coadsorbed on the NaAu2(111) (down) vs Au(221) (up) crystal planes from initial state (IS) via the intermediate complex OOCO* (MS) to the final state (FS) of CO2[45]
2.2 催化加氢
金属间化合物通常由金属元素组成,对空气敏感,因此适用于在惰性或还原气氛下进行的化学反应
多键(如C=C、C≡C、C=O、N=O)化合物的加氢是一类广泛利用金属间化合物催化的探针反应
以碳氧化合物(CO或CO2)加氢为例来评价金属间化合物对双键活化加氢的催化性能
CO或CO2的甲烷化反应常用于煤制天然气的研究
通常,第四周期的过渡金属组成的金属间化合物(MxNy;M=Cu,Ni,Co,Fe;N=Th,U,Si,Ti)[46,47,48]是一类性能良好的储氢材料,因而广泛用于碳氧化合物的催化加氢
例如,Chen等[49]制备了二氧化硅担载的Si-Ni金属间化合物,将其用于CO加氢
作者观察到,与负载单镍催化剂相比,含有金属间化合物Si-Ni晶相(Ni2Si、NiSi或NiSi2)的催化剂显示更高的CO转化率、CH4选择性和更好的催化剂稳定性,如图4所示
作者认为,活性提高的原因可归因于镍和硅之间的强相互作用阻止了纳米粒子的烧结和碳沉积
图4

图4镍基催化剂在空速为48000 mL/(g·h)条件下对CO加氢反应的CO转化率(a)和甲烷选择性(b)[49]
Fig.4(a) CO conversion and (b) CH4 selectivity on the Ni/SiO2 and Si-Ni/SiO2-x (x=250, 350, and 450) catalysts at GHSV=48,000 mL/(g·h) and pressure=1 atm. The short dashed line represented the thermodynamic equilibrium value[49]
除此之外,N?rskov及合作者[50]报道了Ni5Ga3金属间化合物催化剂对CO2加氢制甲醇反应具有很高的催化活性,且与传统的铜/氧化
锌/
氧化铝催化剂相比,Ni5Ga3金属间化合物催化剂上的CO等副产物得率明显下降
除了镍基催化剂以外,Pd基催化剂用于CO2加氢制甲醇反应亦有报道
例如,Ota等[21]制备了Pd2Ga和PdZn纳米金属间化合物催化剂,用于CO2加氢反应制甲醇,发现与单金属相比前者具有更高的转化率与选择性
当然,除了碳氧化合物的加氢反应,金属间化合物对其它加氢反应也表现出良好的催化活性和选择性
例如,用于乙炔半加氢反应的PdGa/Al2O3[51]、Pd2Ga/Al2O3[51]、Pd2Ga/C[52]和PdZn/ZnO[53]具有良好的催化活性,Ga 或Zn 的引入削弱了对目标产物的吸附(即增强了目标产物的脱附)和改善了H2的离解能力,从而提高了目标产物的选择性和得率
用于脂肪炔烃的加氢反应的PdZn/ZnO[54]、Pd3Pb/SiO2[55]和Pd3Bi/SiO2[55]也显示出较高的脂肪烯烃选择性
2.3 水蒸汽重整
在化石能源日渐枯竭、环境污染日益严重的现代社会,氢能等清洁新能源的应用显得尤为重要
甲烷、甲醇和碳氢化合物等燃料的水蒸汽重整是常用的制氢反应,其含氢量高且储运方便,因而广泛用于制氢原料
金属间化合物因其独特的双金属的有序排列,因而对于某些反应显示出高选择性
以甲醇水蒸汽重整(Methanol steam reforming,简称为MSR)反应评价金属间化合物催化剂对C-H键断裂的重整反应的催化性能
最近,Neumann等[56]报道了双金属体系对甲醇水蒸汽重整的催化效果,发现在300oC或390oC条件下利用氢气处理时,PdO/In2O3上会形成PdIn或Pd2In3金属间化合物晶相,这些包含金属间化合物晶相的催化剂在甲醇水蒸汽重整反应中表现出很高的二氧化碳选择性,且在连续反应100 h后仍然保持良好的催化活性
除此之外,体相NiZn金属间化合物同样也表现了极高的二氧化碳选择性[57],催化剂在反应中分解为NiZn、ZnO和Ni0.7Zn0.3,其中Ni0.7Zn0.3为甲醇水蒸汽重整反应的活性中心,对甲醇水蒸汽重整反应表现出极高活性
除了这些金属间化合物以外,类水滑石结构的Pd-Mg-Ga[21]对MSR反应也表现出良好的催化活性,如图5所示
作者认为从Pd-Mg-Ga的类水滑石结构中形成的Pd2Ga金属间化合物为MSR反应的活性中心
图5

图5PdZnAl、PdMgAl和PdMgGa催化剂上MSR反应在250oC时的甲醇反应速率与氢气形成速率[21]
Fig.5Methanol reaction rates and H2 formation rates over the PdZnAl, PdMgGa, and PdMgAl catalysts in the MSR at 250oC[21]
以上是一些金属间化合物催化剂较典型的催化应用
除了这些反应以外,金属间化合物对其它反应也具有优良的催化性能
例如,PtSn[58]和PdZn[59,60]对水煤气变换反应显示良好的催化活性
PdZn在乙烯加氢甲酰化反应中具有高的氧化产物选择性[61]
与单金属Ru相比,Ru2La对催化氨分解为H2和N2的反应表现出更好的催化活性[62]
为了说明金属间化合物作为催化剂所具有的良好催化活性,本文总结了一些较为典型的金属间化合物的制备方法、部分物理性质和催化活性,如表1所示
这些具有优异催化活性的金属间化合物在催化领域具有巨大的应用前景
Table 1
表1
表1文献报道的金属间化合物的合成方法、物理性质和催化活性
Table 1Synthesis methods, physical properties, and catalytic activities of intermetallic compounds reported in the literature
|
Intermetallic compound
|
Synthesis method
|
Crystal phase
|
Particle size
|
Reaction condition
|
Catalytic performance
|
Ref.
|
|
Pd5Ga3
|
Chemical reduction
|
Orthorhomibc
|
5.3 nm
|
0.5% CH4, 4% O2, N2 balance; space velocity (SV): 80000 mL/(g·h)
|
T90% is lower to 372oC, the special reaction rate of Pd is 23.32×10-6 mol/(gPd s) at 290oC.
|
[12]
|
|
Ni3Ga, Ni3Sn2
|
Chemical reduction
|
Cubic
|
3.5~7.5 nm
|
Pretreated with H2; 1 mmol substrate and 0.5 mmol n-dodecane; H2: 500 kPa; 1300 r/min
|
After 13 h continuous reaction, the conversion of various types of alkyne reached more than 90%, and the selectivity of olefins was over 94%.
|
[15]
|
|
PdmMn (M=Ge, In, Sn, Zn)
|
Gas-phase reduction
|
-
|
-
|
12.5% butylene, butylene: O2=1:1, He (balance); total gas flow rate: 120 mL/min
|
PdIn, PdBi or Pd3Fe catalysts show high selectivity for 1,3-Butadiene and 1-butene (more than 50%) and high yield.
|
[20]
|
|
Pd2Ga and PdZn
|
Gas-phase reduction
|
Cubic
|
-
|
H2O/CH3OH=1.0, total gas flow rate: 26 mL/min, methanol concentration: 28.4%
|
PdZnAl exhibited the best catalytic activity, with 87% hydrogen selectivity at 250oC and hydrogen generation rate of 964 μmol/(g·min).
|
[21]
|
|
Ni3Sn, Ni3Sn2, Ni3Sn2, Ni3Sn4
|
Arc melting
|
-
|
25~38 μm
|
The partial pressures of acetylene and hydrogen are 2.7 and 13 kPa, respectively.
|
The acetylene conversion rates of Ni3Sn, Ni3Sn2 and Ni3Sn4 are 2.3×10-6 mol/(g·s), 0.6×10-6 mol/(g·s) and less than 0.001×10-6 mol/(g·s), respectively.
|
[22]
|
|
Pt3Sn
|
Polyol process
|
-
|
5.2±1.0 nm
|
O2/CO=6:1; room temperature
|
The initiation temperature of CO oxidation on Pt3Sn is lower than that on the Pt catalyst.
|
[31]
|
|
Pt3Ti
|
Chemical reduction
|
Cubic
|
2.5 nm
|
2 % CO, 1% O2, 97% He, space velocity (SV): 120000 mL/(g·h)
|
The ignition temperature of CO oxidation on Pt3Ti catalyst is 125oC, which is lower than that on single Pt catalyst.
|
[43]
|
|
Ni2Si, NiSi or NiSi2
|
Chemical vapor deposition
|
Cubic
|
3~4 nm
|
H2/CO=3:1, Ar balance, space velocity (SV): 48000 mL/(g·h)
|
The activity of CO methanation on Ni-Si catalyst is much higher than that on single nickel catalyst, with enhanced stability of nickel sintering resistance at high temperature (500~600oC).
|
[49]
|
|
NiZn
|
Thermal annealing
|
Cubic
|
20~32 μm
|
methanol:H2O=1:1; 0.01 mL/min, N2: 13.2 mL/min, He: 1.6 mL/min
|
NiZn catalyst has good catalytic performance for methanol reforming (80% conversion at 550oC) and good hydrogen selectivity (70%).
|
[57]
|
3 金属间化合物的催化机理
从上述金属间化合物的催化性能中可看出,与单金属催化剂相比,金属间化合物对特定反应的催化活性和选择性均显著的提升,主要是其电子效应、几何效应、空间效应以及原子排列等所致
3.1 电子效应
金属间化合物是由两种及以上金属原子有序排列而成的一类金属化合物,一种元素周围与其它元素存在原子配位,相比于各个单金属,两种金属原子周围的电子云密度均会发生变化
金属间化合物引起特殊的电子效应,而电子状态的剧烈变化会显著影响其对反应物分子的吸附和活化,从而影响其催化活性和选择性
Stamenkovic等[63]发现,Pt3M(M=Ti,V,Fe,Co,Ni)d带中心电位与其氧还原反应(ORR)活性之间存在火山形关系,如图6所示
他们认为,当d带中心电位距离费米能级(火山形曲线的左侧)太远时,O2和中间体的弱吸附限制了反应速率
如果d带中心电位太靠近费米能级(火山形曲线的右侧),则表面物质(如OH)的强吸附会抑制游离Pt的释放,从而限制反应速率
图6

图6在0.1 mol/L HClO4中实验测量的Pt3M表面上ORR在60oC时的比活性与表面d带中心电位之间的关系[63]
Fig.6The relationship of experimentally measured specific activity for the ORR on the Pt3M surface in 0.1 mol/L HClO4 at 60oC with the d-band center position for the Pt-skin surface[63]
Furukawa等[64]也报道了Ni3M金属间化合物(M=Ge,Nb,Sn,Ta,Ti)对氢-氘转换反应的表观活化能与d带中心电位之间的关系,如图7所示
他们发现,氢-氘转换反应的表观活化能与d带中心电位呈现出明显的线性相关性
d带中心电位越低,Ni-H(D)键越弱,从而降低了氢氘的解吸能
对于氢-氘转换反应,解吸能是其反应表观活化能的主要能垒,因而金属间化合物的d带中心电位的改变从根本上影响了催化活性
图7
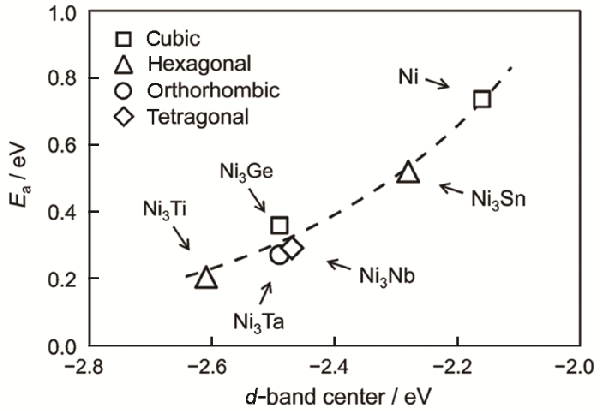
图7Ni和Ni3M金属间化合物对H2-D2平衡反应的表观活化能与d带中心电位的关系[64]
Fig.7A relationship of the apparent activation energies of Ni and Ni3M intermetallic compounds for the H2-D2 equilibrium reaction with their potentials of the d-band centre[64]
3.2 几何效应
金属间化合物中两种金属原子呈现有序排列,即一种金属(N)进入另一种金属(M)的表面晶格
相对于各个单金属,金属间化合物的表面M-M(或N-N)的配位数降低了
对于某些需要多个位点吸附的化学反应,单金属催化剂上M-M(或N-N)的高配位数使得这些吸附或反应较难于进行,原子利用率低,从而影响了其催化活性和选择性
相反,具有较低表面M-M(或N-N)配位数的金属间化合物则不存在此类问题,因而显示较高的催化活性和选择性
特性与第一种金属不同的第二金属的掺入,通常会引起电子结构以及表面吸附强度的急剧变化,从而影响反应的催化活性与选择性,即所谓的金属间化合物所具有的几何效应
Prinz等[65]通过金属间化合物PdGa的(111)和(???1?1?1?)晶面的特殊结构说明了几何效应的影响
图8a所示为PdGa的(111)和(???1?1?1?)晶面的STM照片
(111)晶面上形成三个Pd原子(Pd3)的团簇,而(???1?1?1?)晶面上则仅有单个Pd原子(Pd1)原子
虽然PdGa的(111)和(???1?1?1?)晶面电子结构相似,但其表面的Pd位点不同,存在几何效应
通过CO的吸附实验,作者发现在(111)晶面上随着CO覆盖率的增加,由桥式吸附向线型吸附的迁移,而在(???1?1?1?)晶面上则仅存在线型吸附(图8b)
这种结构与吸附方式的对应关系可归因于几何效应所致
图8

图8(a) PdGa(111)和PdGa(???1?1?1?)晶面的STM照片;(b) PdGa(111)和PdGa(???1?1?1?)晶面上CO吸附的红外谱图[65]
Fig.8(a) STM images of CO adsorbed on Pd3 (A) and Pd1 (B). The atomic surface structure is overlaid on the left-hand site of each image (Pd: cyan, large; Ga: red, small); (b) IR absorption spectra as a function of wavenumber and CO exposure at -183oC on Pd3 (left) and Pd1 (right) [65]
除此以外,Dohyung等[66]研究了有序和无序AuCu双金属纳米粒子对电催化CO2还原的活性,发现有序AuCu纳米粒子能够选择性地还原CO2,主要归因于有序结构有利于析氢,而无序AuCu纳米粒子则相反
这种原子尺度的有序与无序结构也说明了金属间化合物存在几何效应
3.3 空间效应
考虑到金属间化合物表面出现特定且有序的原子排列,调控这些原子排列可能会对吸附物种和反应动力学施加一定的空间限制,从而达到增加催化活性的目的,即所谓的空间效应
其中调控的原子排列会影响一些大分子反应物的吸附态,暴露出某些特定的反应活性位,从而改变产物的选择性
Furukawa等[67]以在RhSb/SiO2上的立体选择性烯烃异构化为模型反应研究了空间效应的影响
研究发现,斜方晶相的Rh和Ru基金属间化合物(RhSb、RuSb和RhGe)在顺式二苯乙烯的异构化中对反式烯烃表现出很高的选择性,相反,立方晶相的Rh和Rh基金属间化合物(RhGa、RhIn和RhZn)则在顺式二苯乙烯的异构化中对反式烯烃表现出较低的选择性,出现过度氢化生成二苯乙烷
除此以外,在顺式-β-甲基苯乙烯的异构化反应中也观察到类似的实验现象
以上结果表明,选择性取决于金属间化合物的空间结构
有趣的是,对于顺式-β-甲基苯乙烯异构化反应,反式烯烃的选择性略低于顺式二苯乙烯异构化反应的产物选择性
对此,作者认为选择性可能不仅取决于金属间化合物的晶相结构,而且还与反应物中烷基的空间位阻有关
因此,金属间化合物的特定表面结构存在一定的空间效应
CO吸附的FT-IR实验表明,RhSb的表面由(211)、(020)和(013)晶面组成(图9)
DFT计算结果表明,表面氢在这些晶面上沿Rh阵列的方向上的扩散受到限制
此外,对吸附的顺式-2-丁烯进行的DFT计算表明,氢接近顺式-2-丁烯的烯基碳原子仅限于一个方向(图9中的浅绿色箭头)
这种独特的氢化行为促进了单原子氢化而产生异构化,但抑制了双原子氢化而形成烷烃
图9
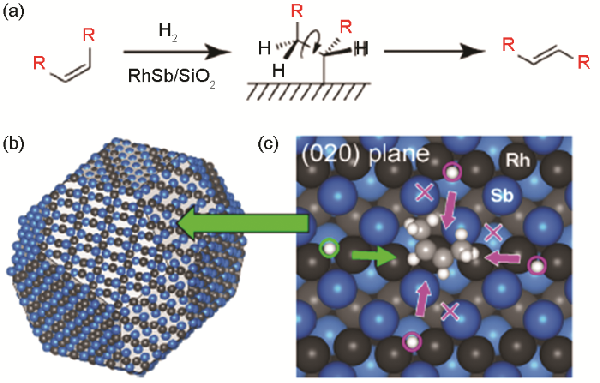
图9(a) 通过烷基中间体的氢控烯烃异构化、(b) 一个RhSb纳米粒子的晶体形状和相应的表面原子排列和(c) 受限氢靠近RhSb (020)晶面上吸附的顺式-2-丁烯[67]
Fig.9(a) Hydrogen-mediated alkene isomerization via alkyl intermediate, (b) equilibrium crystal shape and the corresponding surface atomic arrangement of a RhSb nanoparticle, and (c) limited hydrogen access to cis-2-butene adsorbed on RhSb (020) plane[67]
4 结论与展望
本文总结了金属间化合物的合成方法及其对加氢、氧化、重整等反应的催化性能
与传统的单金属催化剂相比,金属间化合物催化剂对这些反应表现出优异的催化性能
通过关联金属间化合物的催化活性和活性中心可知,电子效应、几何效应、空间效应以及原子排列等因素均可影响其催化性能,特别是金属间化合物的有序原子排列以及特定形貌结构成为影响催化活性的关键因素
另外,金属间化合物具有的特定电子结构(如通过改变第二金属来调控d带中心电位或电子密度)使其对某些反应显示很高的催化活性和选择性
因此,研发新型高效金属间化合物催化材料是一个很有应用潜力的研究方向
除了以上优势之外,金属间化合物在催化过程中还存在一些不容忽视的问题
例如,金属间化合物在反应过程中活性中心不明确,表面结构的稳定性(尤其是含一种还原性很强的金属时金属间化合物在氧化反应中的热稳定性和催化稳定性)有待改善,以及在反应条件下金属间化合物的活性中心之间的协同作用机制尚不清楚
这些问题将会成为今后金属间化合物催化剂的主要研究方向
1 金属间化合物的合成方法1.1 化学还原法1.2 沉积沉淀还原法1.3 电弧熔融法1.4 化学气相沉积法1.5 多元醇法 class="outline_tb" 1005-3093/richHtml_jats1_1/images/img_thumbnail_icon.jpg"/>图22 金属间化合物在催化领域中的应用2.1 氧化反应 class="outline_tb" 1005-3093/richHtml_jats1_1/images/img_thumbnail_icon.jpg"/>图42.3 水蒸汽重整 class="outline_tb" 1005-3093/richHtml_jats1_1/images/table_thumbnail_icon.png"/>表13 金属间化合物的催化机理3.1 电子效应 class="outline_tb" 1005-3093/richHtml_jats1_1/images/img_thumbnail_icon.jpg"/>图73.2 几何效应 class="outline_tb" 1005-3093/richHtml_jats1_1/images/img_thumbnail_icon.jpg"/>图94 结论与展望
声明:
“金属间化合物的合成及其催化应用” 该技术专利(论文)所有权利归属于技术(论文)所有人。仅供学习研究,如用于商业用途,请联系该技术所有人。
我是此专利(论文)的发明人(作者)
 1401
编辑:北方有色网
来源:侯志全,郭萌,刘雨溪,邓积光,戴洪兴
1401
编辑:北方有色网
来源:侯志全,郭萌,刘雨溪,邓积光,戴洪兴



 咨询细节
咨询细节















































































 2026年08月06日 ~ 08日
2026年08月06日 ~ 08日 




















